
令和07/03/01作成
WissenschafftPlus
A new coronavirus linked to human respiratory diseases in China
A new coronavirus linked to human respiratory diseases in China Was a "novel" and "pathogenic" virus found in Wuhan?
A critical review and conclusions
原題名:
「Ein neues Coronavirus im Zusammenhang mit menschlichen Atemwegserkrankungen in China」
邦訳題:
「中国におけるヒト呼吸器疾患に関連する新型コロナウイルス」(HTML)
著者名:
ハンブルグ出身の数学者(匿名希望)
掲載誌:
巻号頁:
No. 4/2021, pp.28-41
掲載年:
2021
Introduction
In the current pandemic, medical professionals, primarily virologists, occupy a prominent position. Their assessments are hardly questioned in our society and are used decisively to justify the measures taken. This approach seems inadequate, especially considering the enormous impact on society as a whole. If we as a society open up a significant space for technocratic thinking and action to shape social life, we must also have the necessary knowledge and the corresponding methodological skills across the board, especially within the ranks of decision-makers. Otherwise, faith comes before knowledge.
This is a problem that is currently becoming dramatically apparent and appears highly toxic. It is now time for many people to come of age, embark on a journey of discovery and trust in their own abilities. It's not about mistrusting, it's about questioning. We cannot achieve or expect absolute truth without doubt, should it exist anywhere. Knowledge must be constantly questioned, tested and dogmas uncovered. These important attributes are the driving force behind scientific work. We should always be open to and grateful for new ideas, arguments and interpretations of the supposedly known, as these enrich our lives like a stimulating elixir.
It can be bolder to doubt the known than to explore the unknown.
Alexander v. Humboldt
This article discusses in detail the scientific publication “A new coronavirus associated with human respiratory disease in China”[1], in which the genetic sequence of the alleged “novel” coronavirus SARS-CoV-2 was proposed for the first time. This paper was published on February 03, 2020 and appeared in the scientific journal Nature.
The analysis is divided into two parts. In the first part, the methodological procedure for constructing the alleged SARS-CoV-2 virus genome is explained. This is followed in the second part by a critical examination of the publication in a scientific sense. Methodological weaknesses, critical questions and control options are pointed out, which question the discovery of a “novel” and “disease-causing” virus in a comprehensible way.
PART A: How was the sequence for the "novel" coronavirus SARS-CoV-2 determined?
The initial situation
A 41-year-old patient who was admitted to the Central Hospital in Wuhan on December 26, 2019 was examined. He was a worker at the local fish market, which was associated as the original outbreak site of the "novel" virus according to epidemiological investigations. The patient was admitted with a temperature of over 37.5 °C, chest tightness, cough, pain and general weakness. Preliminary etiologic investigations ruled out the presence of influenza viruses, among others. Other common respiratory pathogens, such as human adenoviruses, were also ruled out. After the patient's condition did not improve even after the administration of antibiotic and antiviral medication, he was transferred to the intensive care unit and later to another hospital in Wuhan for further treatment.
In order to identify possible pathogens associated with the present disease, bronchoalveolar lavage fluid (BALF) was taken from the patient and subsequently sequenced meta-transcriptomically.
The method section
The scientific publications within virology generally include a methods section in which the procedure is described in more detail. For example, the methodological procedure and the sequencing technologies used are described here and information on the bioinformatic protocols is included.
Patient information and sample collection
A patient with acute onset of fever (temperature above 37.5 ℃), cough and chest tightness who presented to the Central Hospital in Wuhan, China, was examined. During admission, bronchoalveolar lavage fluid (BALF) was collected from the patient and preserved at -80 ℃ until further processing. All patient information, such as examination and laboratory data, was available in the form of clinical records.
The patient sample and RNA sequencing
First, the entire RNA (ribonucleic acid) was extracted from the BALF. Commercial extraction kits are used for this purpose. These kits are generally used in accordance with the manufacturer's instructions. After extraction, the quantity and quality of the RNA solution is assessed before subsequent sequencing. However, before the RNA can be sequenced, a so-called RNA library must be constructed. This involves the technical preparation of the existing RNA for the sequencing step. For example, the RNA must be transcribed into cDNA using reverse transcription and divided into short fragments. In the present case, the ribosomal RNA was removed during library construction according to the manufacturer's instructions. Ribosomal ribonucleic acid (rRNA) is found in the human ribosomes, the site of protein biosynthesis. Before the actual sequencing, the short fragments are amplified using the polymerase chain reaction (PCR) so that the sequencing machine used can detect them and determine the nucleotide sequence. The sequencing of the RNA library was carried out on the MiniSeq platform from Illumina in Shanghai.
| RNA (ribonucleic acid) consists of a phosphate, the sugar ribose and the four bases adenine (A), cytosine (C), guanine (G) and uracil (U). |
| DNA (deoxyribonucleic acid) also consists of a phosphate, the sugar deoxyribose and also four bases, with the base thymine (T) instead of uracil (U). This type of molecule is referred to as a nucleotide and is also designated by the letters A, C, G, T and U. |
| A DNA Sequence is a sequence of nucleotides and is stored in the computer in the form of sequences of letters. |
| The first eight nucleotides of the "spike protein" are: ATGTTTGT [2]. |
Bioinformatic processing of sequence reads and virus identification
Sequencing according to the protocol described above and subsequent adaptation and quality control with the Trimmomatic program resulted in a total of 56,565,928 short sequences (reads) with a length of approximately 150 nucleotides. As explained above, the short sequences can be described with letter sequences consisting of A, T, C, G for the four nucleotides. The letter sequences are stored in ordinary text files. In the present case, the short reads were stored in FASTQ format [3], which contains additional quality information on the individual nucleotides. From this quality information for each individual nucleotide within a short sequence read, the probability that the respective nucleotide was correctly registered by the sequencing machine can be determined.
An attempt is now made to determine longer contiguous nucleotide sequences from the short sequence reads. For this purpose, overlaps between the short sequences are sought. Due to the large number of short reads, in this case around 56 million, bioinformatic programs and powerful computers are required for this step. The longer nucleotide sequences calculated by overlapping are called contigs. The word contig comes from the English word contiguous, which means contiguous. The following diagram illustrates this process.
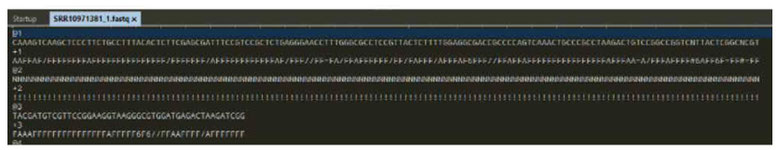
| In addition to the sequence read, quality information for the individual nucleotides (nt) can also be saved in a FASTQ file. |
|
@3 TACGATGTCGTTCCGGAAGGTAAGGGCGTGGATGAGACTAAGATCGG +3 FAAAFFFFFFFFFFFFFFFFFAFFFFF6F6//FFAAFFFF/AFFFFFFF |
|
@ : Identification of the sequence + : Identification of the quality information for the sequence. |
| Target sequence: | Zweifel ist der Motor wissenschaftlichen Arbeitens. |
| Readings: |
Zweifel ist der Motor wissenschaftlichen Arbeitens. Zweifel ist der Motor wissenschaftlichen Arbeitens. |
| Overlap: |
Zweifel ist der Motor wissenschaftlichen Arbeitens. Zweifel ist der Motor wissenschaftlichen Arbeitens. |
| Contigs: | Zweifel ist der Motor wissenschaftlichen Arbeitens. |
| Note that the two contigs found do not cover the entire target sequence. The publications do not usually specify how these gaps are filled. In this case, the German language terms are used and the gap is filled with the syllable "wis", although this was not included in the data. | |
In the publication considered here, the complex contig creation was carried out with the two so-called assemblers Megahit and Trinity in versions v.1.1.3 and v.2.5.1. In order to obtain longer contigs from the millions of short sequence reads, the default settings were used. Prior to this, however, possible sequences of human origin were removed using the human reference genome (human release 32, GRCh38.p13) [4]. After this step, 23,712,657 non-human short sequences remained. The following table gives an overview of the two assemblies.
| Software | Number of contigs | Shortest contig | Longest contig |
|---|---|---|---|
| Megahit (v.1.1.3) | 384,096 | 200 nt | 30,474 nt |
| Trinity (v.2.5.1) | 1,329,960 | 201 nt | 11,760 nt |
Table 1: Overview of results for continuous form production with Megahit and Trinity.
The comparison of the contigs with the known sequences in the relevant nucleotide and protein databases revealed that the longest contigs (Megahit: 30,474 nt and Trinity 11,760 nt) were very similar to the SARS-like bat virus SL-CoVZC45, MG772933. According to the authors, the longer of the two contigs, with a length of 30,474 nucleotides, covered almost the entire virus genome and was used for primer design for subsequent PCR confirmation and determination of the genome termini. The viral genome was organized by sequence alignment using two representative betacoronaviruses, namely SARS-CoV Tor2, AY274119 and bat SL-CoVZC45, MG772933. The former is associated with humans and the latter with bats.
Data availability
The sequence reads were deposited in the NCBI Sequence Read Archive (SRA) database under the BioProject accession number PRJNA603194. The complete genome sequence of WHCV (now SARS-CoV-2) has been deposited in GenBank under the identifier MN908947.
Control attempts
No control tests are documented in this publication.
PART B: Critical review of the methods and conclusions
After the procedure for determining the viral genome of the alleged "novel" and "pathogenic" SARS-CoV-2 virus has been examined in detail, this section takes a critical look at the individual work steps. Questioning scientific findings is the elixir of living science.
Notes: Data reporting
The scientists involved were not blind and therefore had knowledge of the patient's medical history and symptoms. This probably influenced the authors to look for possible respiratory pathogens. Already at this point there would be a simple control option.
| 1st control option: A second research team examines the same patient sample without information about the patient, such as their origin and medical history or their clinical symptoms. |
| Control objective: Do two independent research groups obtain the identical claimed virus genome without knowing the patient's clinical picture? |
Notes:
The patient sample and RNA sequencing
As explained in more detail in the section "The patient sample and RNA sequencing", a number of technical steps are necessary before the actual sequencing, i.e. the determination of the nucleotide sequence of short fragments, can be carried out.
For example, commercial kits are used
- to extract total RNA from the sample,
- to remove ribosomal RNA from the sample,
- to convert RNA into cDNA,
- to fragment the RNA or cDNA into short pieces, or
- for the amplification (PCR) of the cDNA in order to be able to determine the nucleotide sequence.
Commercial technical equipment is used to check the quantity and quality of the RNA. These are complex processes that can only be controlled to a very limited extent by the scientists. This leads directly to a further control option that is not documented in the scientific literature.
| 2nd control option: The initial sample is prepared for the actual sequencing step using a different protocol. |
| Control objective: Different protocols for the preparation of the patient sample lead to short sequence reads from which identical or sufficiently similar contigs can be generated with the assemblers used. |
It is therefore noted that the sequencing of RNA from the available bronchoalveolar lavage fluid requires a highly complex protocol. The steps involved are difficult to control. It is therefore questionable to what extent the reproducibility required for scientific validity can be guaranteed without knowledge of a reference genome.
Notes: Bioinformatic processing of sequence reads and virus identificationFirst of all, the clear difference between the results of the two assemblers Megahit and Trinitiy is striking.
The discrepancy between the two results is enormous. For example, the longest contiguous sequence found by overlapping with Megahit was 30,474 nucleotides, while Trinitiy generated a longest contig of 11,760 nucleotides from the same data set. Conversely, Trinitiy produced significantly more contiguous sequence pieces, namely 1,329,960 pieces, than Megahit (384,096). This immediately raises the question:
What would the sequence proposal for the alleged SARS-CoV-2 virus have been if the Megahit software and the corresponding reference genomes (bat-like SARS viruses and previously known SARS viruses associated with humans) had not existed?
Another important question is:
Why is the length of the claimed coronavirus SARS-CoV-2 only 29,903 nucleotides, which is 571 nucleotides less than the longest contig, which comprises 30,474 nucleotides?
The big difference between the two software programs in terms of the maximum contig length is extremely remarkable. There do not appear to be any recognized regularity criteria for the algorithms used to find overlaps. This is particularly critical from a scientific point of view, especially with regard to reproducibility.
Furthermore, the analysis to date, particularly with regard to possible random errors during the sequencing step or the discovery of nucleotide sequences that may not exist in reality, shows that the calculated sequences or claimed virus genomes can only be models for elementary logical reasons. It is therefore not permissible to speak of a true virus genome or a true virus sequence. This important fact and possible procedures for model validation are not mentioned in the scientific literature, or only in passing.
In "Choice of assemblers has a critical impact on de novo assembly of SARS-CoV-2 genome and characterizing variants" [5], the authors show that the choice of assembler plays a significant role in the reconstruction of the SARS-CoV-2 genome from several hundred samples. Furthermore, they showed that at least 9% of the variants between Megahit and metaSPAdes (another assembler) are unique to the assembly methods. The scientists conclude that their analyses show the crucial role of the assembly methods used for the construction of SARS-CoV-2 genomes from short reads on the characterization of so-called variants.
This observation leads to the following question, among others:
What exactly are virus variants or virus mutations? Biological reality or a bioinformatic artifact?
In summary, it can be said that assembly results show large differences.
This publication mentions that the longest contiguous sequence computed with Megahit (30,474 nt) covers almost the entire viral genome. PCR primers were designed on the basis of this sequence. In the supplementary Table 8. "PCR primers used in this study", 52 primers for genome amplification are listed. These cover the entire purported viral genome evenly, as shown in the following graph.
![Figure 1: Coverage of nucleotides of SARS-CoV-2 with short reads, distribution of the 52 primers for whole genome amplification and 99% interval. The nucleotide coverage was determined using Bowtie2 [6] and Samtools [7].](https://image.jimcdn.com/app/cms/image/transf/dimension=778x10000:format=jpg/path/sdde7d3e553b4a76d/image/ic99b45a9605c2060/version/1740825419/image.jpg)
It is not clear from this publication exactly how the PCR confirmation or the determination of the genome terms was carried out. However, Figure 1 shows a particularly high coverage of the nucleotides of the claimed virus genome in the vicinity of the listed primers. The strongly varying depth of coverage of the individual nucleotide positions is also striking. Assuming that all 29,903 nucleotide positions occur with equal probability in SARS-CoV-2 associated reads, the coverage for each nucleotide position should lie between the two lines with 99% probability. This is not the case for about 90% of nucleotide positions, which is quite remarkable. A priori, one would expect that if sufficient viral RNA is present in the sample and a sufficient number of sequence fragments are read, a homogeneous coverage of the nucleotides within the viral genome would be achieved. Furthermore, the question arises as to why a second, specific primer-based confirmation and determination step is necessary to clarify the genome terms. This leads to the assumption that this step leads to the distortion clearly recognizable in the diagram. Furthermore, the impression could arise that the use of the many PCR primers specific for the claimed virus genome and the amplification with an unscientifically high number of 35 cycles results in "connection sequences" that do not exist in reality.
This assumption is supported by the sequencing of the entire RNA of a commercial and not "infected" culture of human cells. The necessary amplification of the cDNA fragments was carried out by polymerase chain reaction (PCR) with 14 cycles and exclusively using random hexamers. This means short primers with 6 random and unspecific nucleotides, which should lead to a relatively homogeneous coverage of the calculated contigs by the associated sequence reads.
![Figure 2: Coverage of a contig (21,814 nt) by its associated sequence reads from human RNA and 99% interval. Nucleotide coverage was determined using Bowtie2 [6] and Samtools [7].](https://image.jimcdn.com/app/cms/image/transf/dimension=778x10000:format=jpg/path/sdde7d3e553b4a76d/image/i2fe30eed39d27d0f/version/1740864206/image.jpg)
In contrast to Figure 1, Figure 2 shows a much more homogeneous coverage of the 21,814 nucleotide positions, even though the coverage for many nucleotide positions is outside the 99% interval. However, the deviations here are rather undistorted below and above the 99% interval.
| The contig of length 21,814 nt considered in Figure 2 was calculated from the RNA of a commercial human and non-"infected" cell culture using the assembler Megahit. A comparison using Blastn with the entire nucleotide database showed a high match (99.91%), over almost the entire contig length, with the mRNA sequence: Homo sapiens spectrin repeat containing nuclear envelope protein 2 (SYNE2), transcript variant 5, mRNA, Accession: NM_182914. |
| In the corresponding publication entitled "A novel SYNE2 mutation identified by whole exome sequencing in a Korean family with Emery-Dreifuss muscular dystrophy" [8], it says under Rusultate [... novel de novo pathogenic heterozygous missense mutation (NM_182914.2: c.4858G > A; p.Ala1620Thr) of the SYNE2 gene, which had not been previously reported was identified by whole exome sequencing in the proband and by Sanger sequencing in his son. ...]. |
| An interesting observation in itself! |
Regardless of this observation, it can be stated that the aforementioned mRNA sequence with the identifier NM_182914.2 could be assembled with readings of the RNA fragments of a non-“infected” cell culture of human origin, which were amplified using random and thus unspecific hexamers with only 14 cycles before the actual sequencing. It was not necessary to perform a PCR confirmation or determination using specific primers or to use any reference sequences. This raises the following natural questions.
Why does the longest contig not already contain the entire viral genome sequence of SARS-CoV-2? Why are a large number of specific primer and reference sequences required for the final equence determination?
Why is the longest contig (30,474 nt) longer than the claimed viral sequence of SARS-CoV-2 (29,903 nt)?
First of all, this contradicts naive logic. If 30,474 contiguous nucleotides are found, then the "true" genome sequence should contain at least 30,474 nucleotides. Otherwise, some of the overlaps identified by Megahit would be declared false. No standards for this are documented in the scientific literature.
In the following, simple control experiments will be presented that require only basic knowledge of the Linux operating system, a certain amount of familiarization with the bioinformatics computer programs used and the published sequence data.
| 1st control trial: Does the record under BioProject accession number PRJNA603194, 56,565,928 contain short reads. |
|
Protocol: Download the sequence data: fastq-dump --split-files --origfmt --gzip SRR10971381 |
Result: The sequences can be downloaded in FASTQ format using the "fastq-dump" command of the SRA Toolkit package [9] and the paired-end reads can be conveniently saved in two
text files.
Each of the two text files contains 28,282,964 short sequences with an average length of around 142 bp. In total, therefore, both files contain 56,565,928 short sequences. However, it is striking
that a significant number of the sequences consist of N's, i.e. unknown nucleotides. These could be erroneous reads or subsequently overwritten sequence reads, such as human sequence reads. In
terms of scientific reproducibility, this procedure should be considered critical.
| 2nd control test: Can the assemblies be repeated with the published sequences? In particular, can the longest contigs be reproduced with Megahit and Trinitiy? |
|
Protocol: Preparation of the data such as adapter removal or elimination of reads of poor quality with the fastp software [10]: fastp -i SRR10971381_1.fastq.gz -I SRR10971381_2.fastq.gz -o SRR10971381_1.fastq -O SRR10971381_2.fastq |
| Assembly with Megahit v.1.2.9 [11] (default settings): megahit -1 SRR10971381_1.fastq -2 SRR10971381_2.fastq -o megahit_result |
| Assembly with Trinity v.2.5.1 [12] (default settings): Trinity --seqType fa --left SRR10971381_1. fa --right SRR10971381_2.fa --CPU 6 --max_memory 20G |
Remarks: First of all, it should be mentioned that the Megahit software version v.1.1.3 does not run on the computers used here. Therefore, the current version v.1.2.9 was used. The Trinity software is not compatible with FASTQ files. Trinity was therefore used with the setting -SeqType fa (FASTA). In contrast to FASTQ files, FASTA files only contain the sequences without quality information on the individual bases. These circumstances must be taken into account when analyzing the results.
Result: The following table provides a summarized overview of the results.
| Software | Number of contigs | Shortest contig | Longest contig |
|---|---|---|---|
| Megahit (v.1.2.9) | 28,459* | 200 nt | 29,802* nt |
| Trinity (v.2.5.1) | 157,283 | 201 nt | 29,875 nt |
Table 2: Assembly results of the 2nd control experiment
*The assembly without using the fastp software with the published sequences results in a total number of contigs of 29,463 and a longest contig of 29,802 bp.
The second control test shows an unexpected result. Both the total number of contigs and the lengths of the maximum contiguous sequences differ significantly from the published results. Also noteworthy is the observation that a longer contiguous sequence can be calculated with Trinitiy than specified in the publication [1]. Furthermore, the longest contigs match almost completely with the published genome "MN908947", as a blastn query with the standard nucleotide database shows.
This means that the two longest contigs cannot be reproduced!
The published sequence data can therefore not be the original, raw sequence data. However, it is noteworthy that the total number of sequences provided (56,565,928) corresponds to the information in the publication in question.
As described above, RNA sequences of human origin were eliminated. The human reference genome (human release 32, GRCh38.p13) was used for this purpose. Wikipedia [4] states [... The human reference genome is derived from thirteen anonymous volunteers from Buffalo, New York. Donors were recruited by advertisement in The Buffalo News, on Sunday, March 23, 1997. ...]. To what extent is the reference genome specific to humans and therefore suitable for the reliable detection of RNA sequences of human origin? This question can be answered with the next control experiment, which follows on naturally from control experiment 2.
| 3rd control experiment: Do the contiguous sequences calculated in the 2nd control experiment with Megahit (v.1.2.9) contain sequences of human origin? |
|
Protocol: Selection of the 25 longest contigs and comparison with the NCBI nucleotide database |
| Blastn: blastn -db nt -remote -query $Input.fasta -out $Result.txt -outfmt "6 qseqid sseqid stitle pident length mismatch gapopen qstart qend sstart send evalue bitscore" |
Result: The following table shows selected query hits. The longest contig calculated with Megahit (k141_11881) shows a perfect match with the published sequence for SARS-CoV-2 over a length of 29,801 nucleotides. The two contigs (k141_18050 and k141_13168) show a high concordance with human sequences over a length of up to about 6,000 nucleotides.

According to the subject descriptions, these are ribosomal and messenger RNA (rRNA and mRNA). This would mean that not all human RNA sequences were fully removed from the original sequences.
Finally, it should be noted that the two longest contigs (Megahit: 30,474 nt and Trinity: 11,760 nt) show a high nucleotide-by-nucleotide match with the bat corona virus bat SL-CoVZC45, MG772933 of 89.1% and 90.4%, respectively. However, it is not stated how many nucleotides match in total. It is quite possible that only 10,000 nucleotides of the total of 30,474 nucleotides match 89.1% in the case of Megahit. This would mean that 20,474 nucleotides would have no significant match with bat SL-CoVZC45.
Note: Control tests
As already stated, no control tests were documented in the publication under review. It must therefore be assumed that no control tests were carried out. The following are other obvious control options that could have corroborated the findings.
| 3rd control option: Can the claimed virus genome also be obtained from samples of other cases of disease with an identical symptom complex, but not from samples of healthy individuals. |
| Control objective: To confirm the assumption that the presence of the calculated sequence is causally responsible for a very specific symptom complex. |
| 4th control option: Can the claimed virus genome also be obtained from RNA sources that are known to contain no pathogenic “viruses”? |
| Control objective: Are the RNA fragments used to assemble the claimed SARS-CoV-2 genome of viral origin? |
Summary and conclusion
The entire RNA was extracted from the patient sample of a case of the disease in Wuhan. According to the authors, human RNA fragments were removed. Control experiment 3 has shown that the published sequences for the claimed SARS-CoV-2 virus genome with the identification MN908947 most likely still contain ribosomal or messenger RNA of human origin.
The 2nd control experiment has shown that the published results are not reproducible. Rather, the published sequences cannot match the original sequences. According to the description in this publication, it could not be assumed that both Megahit and Trinitiy can be used to obtain almost the complete "virus genome". This observation is a cause for concern, as it is no longer possible to verify to what extent the 30,474 nt contig assembled with Megahit matches the bat coronavirus bat SL-CoVZC45 and to what extent this deviates from the final sequence proposal for SARS-CoV-2.
No control tests were documented. It can therefore be assumed that no control tests were carried out. Possible control experiments were outlined in this article, although the list is not exhaustive.
It is a priori completely unclear whether the disease of the patient in question is of "viral" origin. Furthermore, it could be shown that the origin of the RNA fragments used to construct the alleged viral genome was not determined. It could simply be RNA of human origin. The question also arises as to whether it is even possible to clarify the origin with the methods used here. In short: Are the sequences used of "viral" origin or not?
This question inevitably leads to the concept of virus isolation, in the sense of the word "isolation". In the present case, no (virus) particle was found. The naive protocol for finding a possible (virus) particle is as follows:
- Discover “many” particles of the same morphology in the patient sample (here: bronchoalveolar lavage fluid). These should be there, since virologists assign the claimed SARS-CoV-2 virus
particles a size in the range of about 100 nm.
- Separate these particles from everything else.
- Take several different samples of the particles found.
- Sequence all samples independently of each other.
- Get (almost) the same sequence in all cases.
Then, in the scientific sense, a particle characterized by its sequence and morphology would have been described. However, this would by no means be proof of the possible pathogenicity or transmissibility of this particle.
In conclusion, it should therefore be noted: The publication "A new coronavirus associated with human respiratory disease in China" [1] did not prove the existence of a "novel virus" or a novel "virus sequence" or a pathogenic agent. In particular, no causal cause was found for the clinical symptom complex of the examined patient.
References
[1] Fan Wu u. a. „A new coronavirus associated with
human respiratory disease in China“. In: Nature 579.7798 (2020), S. 265-269. DOI: 10.1038/s41586-020-2008-3.
[2] Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, co - Nucleotide - NCBI. URL: https://www.ncbi.nlm.nih.gov/nuccore/MN908947.3.
[3] FASTQ format. Aug. 2021. URL: https://en.wikipedia.org/wiki/FASTQ_format.
[4] Reference genome. Sep. 2021. URL: https://en.wikipedia.org/wiki/Reference_genome.
[5] Rashedul Islam u. a. „Choice of assemblers has a critical impact on de novo assembly of SARS-CoV-2 genome and
characterizing variants“. In: Briefings in Bioinformatics 22.5 (2021). DOI: 10.1093/bib/bbab102.
[6] Bowtie 2. URL: http://bowtie-bio.sourceforge.net/bowtie2/index.shtml.
[7] Samtools. samtools/samtools: Tools (written in C using htslib) for manipulating next-generation sequencing data. URL: https://github.com/sam-tools/samtools.
[8] Sook Joung Lee u. a. „A novel SYNE2 mutation identified by whole exome sequencing in a Korean family with Emery-Dreifuss muscular dystrophy“. In:Clinica Chimica Acta
506 (2020), S. 50-54. DOI:10.1016/j.cca.2020.03.021.
[9] Ncbi. ncbi/sra-tools: SRA Tools. URL: https://github.com/ncbi/sra-tools.
[10] OpenGene. OpenGene/fastp: An ultra-fast all-in-one FASTQ preprocessor (QC/adapters/trimming/_ltering/splitting/merging...) URL: https://github.com/OpenGene/fastp.
[11] Voutcn. voutcn/megahit: Ultra-fast and memory-efficient (meta-)genome assembler. URL: https://github.com/voutcn/megahit.
[12] Trinityrnaseq. trinityrnaseq/trinityrnaseq: Trinity RNA-Seq de novo transcriptome assembly. URL: https://github.com/trinityrnaseq/trinityrna-seq.