
2-1節
アセンブリ関係用語説明
この第2章を書くに当たって、そのような流れで書くのかは大体決めていましたが、今回ある論文と出会ってその内容を理解するために遺伝子配列決定について勉強したら、この分野に対してかなり勘違いしていることが分かったので、基本に戻ることにしました。
次節では、上述の論文にてついて書きます。
その内容の理解を助けるため、そして私の勉強のため、遺伝子配列決定について基本的な説明をします。
次世代シーケンサー
遺伝子配列決定において仮想化技術を探るのに、次世代シーケンサーについて詳しく知る必要はありませんでした。
右に示すような装置を用いて行うのは、遺伝子配列を読み取って、そのデータをテキストファイルに変換することです。
ただし、次世代シーケンサーが病原性ウイルスの仮想化には大きく貢献していることは間違いないでしょう。
何故なら、旧世代のシーケンサーであれば、単離されていない遺伝子物質の遺伝子配列を決定するのは困難だからです。
次世代シーケンサーは同時並列に複数の遺伝子物質の遺伝子配列を読み込むことができますので、様々な遺伝子物質を含んだ検体からも簡単に遺伝子配列を読み込むことが可能です。
問題は、その関係ない物質同士の遺伝子配列をどのようにして繋いで、それらしい遺伝子配列を作るかです。
リード
旧世代でも次世代でもシーケンサーでは、対象の遺伝子物質を最初から最後まで読むことはできません。
ですから、目的の遺伝子物質を適当な長さに切り刻みます。
切り刻んだ部分の遺伝子配列のことを「リード」と言います。
ただ、1本の遺伝子物質を切り刻んでも、前後関係が分かりません。
そこで、対象の遺伝子物質をたくさんコピーを作っておいて、切り刻んでやります。
そうすると重なる部分ができますので、重なる部分を「のりしろ」にしてリードをつないでいきます。

もの凄く簡略的に説明しますが、例えば目的の遺伝子物質を適当な長さで切り刻んだ後、次世代シーケンサーによって、左図のようなリードが7本得られたとしましょう。
これをそれぞれ重なる部分を探して、順番に並べてやります。
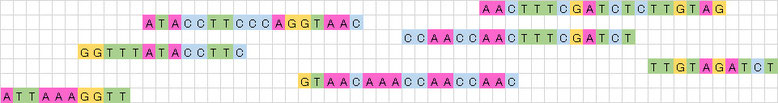
これを左の先頭から順に一本の遺伝子配列にしてやると、下図のようになります。

上図で赤く塗られた部分が重なった部分、すなわち「のりしろ」の部分です。
青く塗られた部分が重なりがなかった部分です。
実際は、もっと大量に同じようなリードが存在していますので、均質に切り刻んでいれば重ならない部分はありません。
アセンブラ
次世代シーケンサーで得られた大量のリードのテキストデータから対象となる遺伝子物質の遺伝子配列を決定するのは、「アセンブラ」と呼ばれるソフトウェアです。
アセンブラは次世代シーケンサー上ではなくコンピューターで動かします。
フリーソフトで配付されているのもあり、皆さんのコンピューターでも動かすことができます。
FASTQ形式
上図の説明のように、リードのデータはA、G 、T、Cの塩基の並びの情報だけでなく、各塩基の確からしさの情報も含んでいる。
この品質情報は精度の高い遺伝子配列決定のために利用されます。
具体的には以下のような形式になります。
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36 GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC + IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC @SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=36 GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGA + IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBI
行の先頭が「@」である行は配列IDおよび配列の説明です。
その下の行が塩基配列です。
3行目の先頭は「+」です。
その下の行が「クオリティ値」で、2行目の塩基の品質を表しています。
そして、この4行で一つのリードを意味していて、リードの数だけこの4行が繰り返されます。
FASTA形式
ついでにFASTA形式も説明しておく。
これはFASTQ形式の塩基配列の情報とIDの情報しかない形式です。
>SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36 GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC >SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=36 GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGA
前処理
アセンブラが対応している形式を使ってアセンブル、すなわち遺伝子配列決定を行うわけですが、そのまま使うのではなくて、アセンブルしやすいように前処理をしてやります。
前処理は仮想化と関係ないので、詳細は省きます。
コンティグ
アセンブラのアルゴリズムによって、リードを組み合わせていくわけですが、組み合わせてできた遺伝子配列のことを「コンティグ」と言います。
通常、可能な組み合わせの数だけコンティグを大量に生成します。
理屈から考えれば、単離されている遺伝子物質からのアセンブリであれば、最大長のコンティグが求めたい遺伝子配列になります。
一方、単離されていない様々な遺伝子物質が含まれている検体からのアセンブリであれば、それぞれのコンティグが検体に含まれている遺伝子物質の遺伝子配列であると考えられます。
カバレッジ
カバレッジとは、得られた遺伝子配列の中の1つの塩基が何回重ねて読まれたかという指標です。

上図で、一番上の遺伝子配列が得られた遺伝子配列です。
リファレンス配列とも言います。
この遺伝子配列をアセンブルするときに利用したリードを並べていったときに、のりしろになる部分が何回も重ねられているのが分かります。
その「重ねられている回数」がカバレッジです。
上図ではリファレンス配列の赤い丸がついたところの塩基(C:シトシン)の部分は20回重ねられているので、「20×カバレッジ」と言います。

